NicheSphere differential co-localization tutorial
NicheSphere is an sc-verse compatible Python library which allows the user to find differentially co-localized cellular niches and biological processes involved in their interactions based on cell type pairs co-localization probabilities in different conditions. Cell type pair co-localization probabilities are obtained in different ways: from deconvoluted Visium 10x / PIC-seq data (probabilities of finding each cell type in each spot / multiplet), or counting cell boundaries overlaps for each cell type pair in single cell spatial data (MERFISH , CODEX …). This tutorial focuses on defining groups of cells that converge or split in disease (Ischemia) based on differential co-localization.
NicheSphere also offers the possibility to look at localized differential cell - cell communication based on Ligand-Receptor pairs expression data. This is addressed in the localized differential communication tutorial.
1. Libraries and functions
[1]:
import pandas as pd
import scipy
import seaborn as sns
import matplotlib.pyplot as plt
import matplotlib.colors as mcolors
import networkx as nx
import warnings
import scanpy as sc
import mudata as md
import numpy as np
from community_layout.layout_class import CommunityLayout
warnings.filterwarnings("ignore")
import nichesphere
COMMUNITY LAYOUT: Datashader not found, edge bundling not available
2. Data at first glance
In this example we will use data from the Myocardial Infarction atlas from Kuppe, C. et. Al., 2022
[2]:
mudata=md.read('heart_MI_ST_SC_23samples.h5mu')
mudata
[2]:
MuData object with n_obs × n_vars = 206792 × 39120
2 modalities
visium: 73904 x 11704
obs: 'n_counts', 'n_genes', 'percent.mt', 'Adipocyte', 'Cardiomyocyte', 'Endothelial', 'Fibroblast', 'Lymphoid', 'Mast', 'Myeloid', 'Neuronal', 'Pericyte', 'Cycling.cells', 'vSMCs', 'cell_type_original', 'assay_ontology_term_id', 'cell_type_ontology_term_id', 'development_stage_ontology_term_id', 'disease_ontology_term_id', 'ethnicity_ontology_term_id', 'is_primary_data', 'organism_ontology_term_id', 'sex_ontology_term_id', 'tissue_ontology_term_id', 'patient_region_id', 'orig_ident', 'batch'
var: 'features'
obsm: 'X_pca', 'X_spatial', 'X_umap'
sc: 132888 x 27416
obs: 'orig_ident', 'nCount_RNA', 'nFeature_RNA', 'percent_mt', 'doublet_score', 'doublet', 'dissociation_s1', 'opt_clust', 'patient', 'batch', 'opt_clust_integrated', 'cell_type', 'ident', 'nFeaturess_RNA', 'cell_subtype', 'cell_subtype_available', 'cell_subtype2', 'patient_region_id', 'patient_group', 'sampleType'
var: 'n_counts'
obsm: 'X_harmony', 'X_pca', 'X_umap_harmony'This is a subset with 23 samples (samples with less than 1500 cells in the snRNA-seq data were filtered out), and 33 different cell subtypes
[3]:
mudata['sc'].obsm['umap']=mudata['sc'].obsm['X_umap_harmony']
sc.pl.umap(mudata['sc'],
color=['cell_type', 'cell_subtype2'], wspace=0.3)

In this case, we will get cell type co-localization probabilities from deconvoluted Visium data (Cell type probabilities per spot):
In a previous step, we used MOSCOT(Klein et. al., 2025) to deconvolute cell subtypes in visium slices from the same 23 samples , getting matrices of probabilities of each cell being in each spot. Then we got cell type probabilities per spot summing the probabilities of cells of the same kind in each spot; thus getting cell type probability matrices for all samples.
(you can have a closer look at these steps in the preprocessing tutorial)
[4]:
CTprops=pd.read_csv('CTprops.csv', index_col=0)
CTprops.head()
[4]:
| Fib1_SCARA5 | damaged_CM | Capillary_Endo | LYVE_FOLR_Macrophages | Fib3_C7 | healthy_CM | Fib2_Myofib | Endocardial_Endo | Arterial_Endo | Neuronal | ... | CCL18_Macrophages | perivascular_fibroblasts | CD_4 | vSMCs_2 | Lymphatic_Endo | NK | CD_8 | Purkinje_fibers | Adipo | NK_T | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AAACAAGTATCTCCCA-1-1-0-0-0 | 0.000000e+00 | 0.000000e+00 | 0.0 | 8.333133e-16 | 0.000000 | 0.000000e+00 | 0.428865 | 0.000000 | 0.0 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | NaN | NaN |
| AAACAATCTACTAGCA-1-1-0-0-0 | 0.000000e+00 | 2.691729e-21 | 0.0 | 0.000000e+00 | 0.445912 | 5.540884e-01 | 0.000000 | 0.000000 | 0.0 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | NaN | NaN |
| AAACACCAATAACTGC-1-1-0-0-0 | 0.000000e+00 | 0.000000e+00 | 0.0 | 0.000000e+00 | 0.000000 | 0.000000e+00 | 0.000000 | 0.000030 | 0.0 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.499508 | 0.0 | 0.0 | NaN | NaN |
| AAACAGAGCGACTCCT-1-1-0-0-0 | 1.373226e-25 | 0.000000e+00 | 0.0 | 0.000000e+00 | 0.499762 | 3.111796e-13 | 0.500238 | 0.000000 | 0.0 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | NaN | NaN |
| AAACAGCTTTCAGAAG-1-1-0-0-0 | 0.000000e+00 | 0.000000e+00 | 0.0 | 0.000000e+00 | 0.000000 | 0.000000e+00 | 0.443230 | 0.113118 | 0.0 | 0.0 | ... | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 | 0.0 | NaN | NaN |
5 rows × 33 columns
From these deconvolution results, we can look at cell type proportions per sample. For this we will need the spot ID and sample correspondence:
[5]:
spotSamples=mudata['visium'].obs.patient_region_id
spotSamples.reset_index().head()
[5]:
| index | patient_region_id | |
|---|---|---|
| 0 | AAACAAGTATCTCCCA-1-0-0-0-0-0-0-0-0-0-0-0-0-0-0... | control_P17 |
| 1 | AAACACCAATAACTGC-1-0-0-0-0-0-0-0-0-0-0-0-0-0-0... | control_P17 |
| 2 | AAACAGCTTTCAGAAG-1-0-0-0-0-0-0-0-0-0-0-0-0-0-0... | control_P17 |
| 3 | AAACAGGGTCTATATT-1-0-0-0-0-0-0-0-0-0-0-0-0-0-0... | control_P17 |
| 4 | AAACCGGGTAGGTACC-1-0-0-0-0-0-0-0-0-0-0-0-0-0-0... | control_P17 |
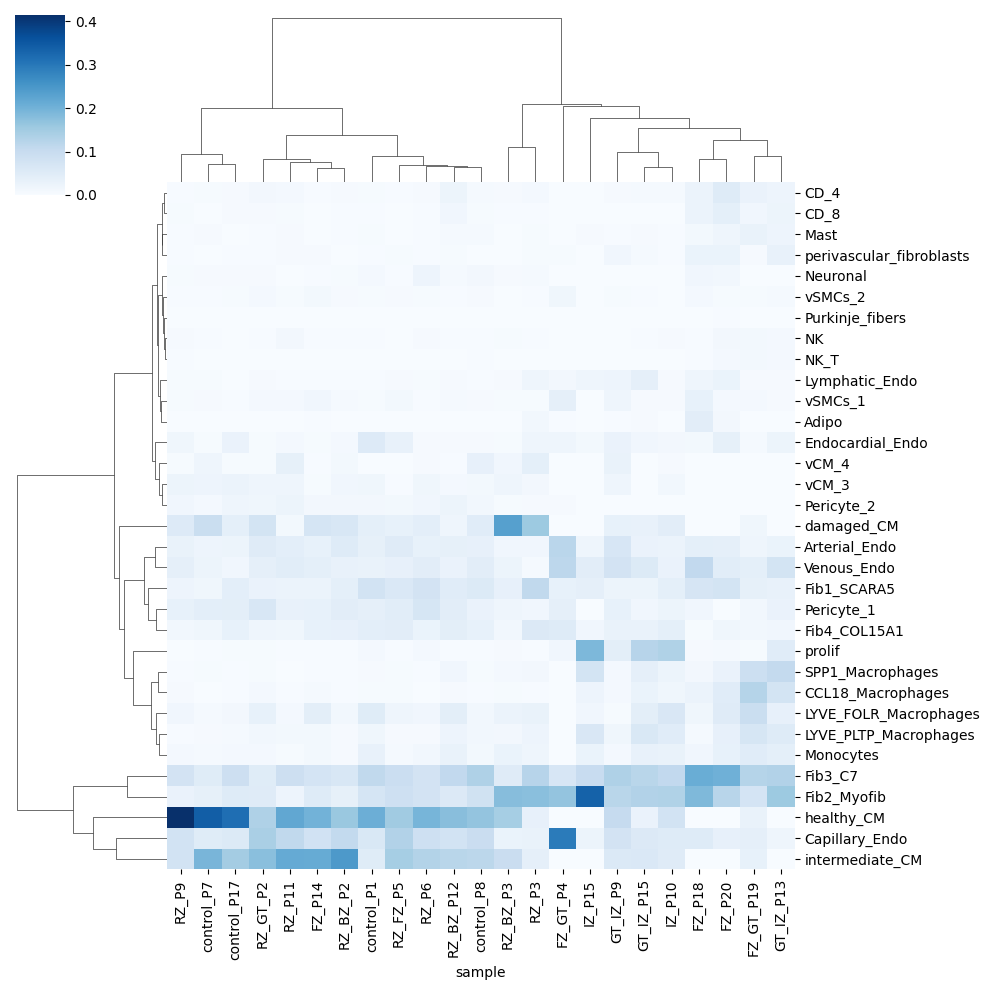
A way to check the deconvolution proportions is using a clustermap
[6]:
CTprops_sample=CTprops.copy()
CTprops_sample['sample']=mudata['visium'].obs.patient_region_id
sns.clustermap(CTprops_sample.groupby('sample').sum().T/CTprops_sample.groupby('sample').sum().sum(axis=1) ,
cmap='Blues', method='ward')
[6]:
<seaborn.matrix.ClusterGrid at 0x15536a4c0590>
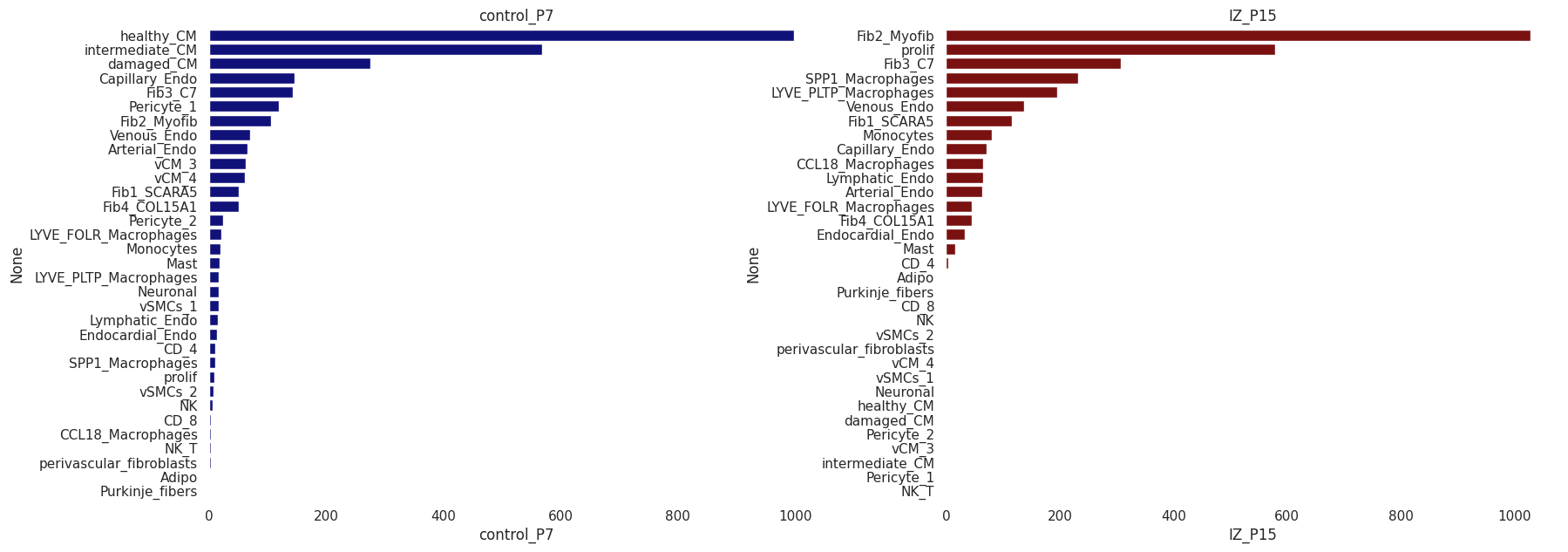
Alternativelly, we can check the deconvolution proportions using barplots
[7]:
t1=pd.DataFrame(CTprops[spotSamples=='control_P7'].sum(), columns=['control_P7'])
t2=pd.DataFrame(CTprops[spotSamples=='IZ_P15'].sum(), columns=['IZ_P15'])
[8]:
sns.set(font_scale=1)
sns.set_style(rc = {'axes.facecolor': 'white'})
fig, axes = plt.subplots(1, 2, figsize=(20, 7))
sns.barplot(ax=axes[0], y=t1.sort_values('control_P7', ascending=False).index, x='control_P7',
data=t1.sort_values('control_P7', ascending=False), color='darkblue')
axes[0].set_title('control_P7')
sns.barplot(ax=axes[1], y=t2.sort_values('IZ_P15', ascending=False).index, x='IZ_P15',
data=t2.sort_values('IZ_P15', ascending=False), color='darkred')
axes[1].set_title('IZ_P15')
[8]:
Text(0.5, 1.0, 'IZ_P15')
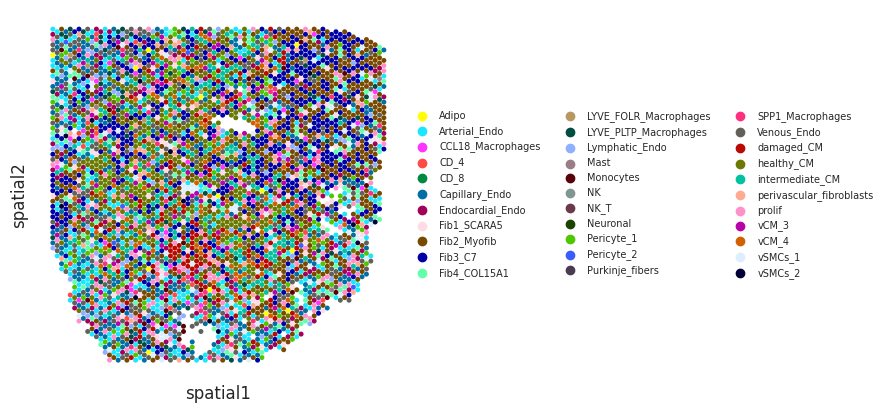
We can visualize cell type deconvolution results in slices (spots are colored by the the cell type with highest proportion). For this we will need the spatial coordinates of the spots in the Visium slices need to be in the slot uns[‘spatial’] of the Visium anndata object:
[9]:
mudata['visium'].uns['spatial']=mudata['visium'].obsm['X_spatial']
[10]:
idPat = 'GT_IZ_P9'
nichesphere.coloc.spatialCTPlot(adata=mudata['visium'][mudata['visium'].obs.patient_region_id==idPat].copy(),
CTprobs=CTprops.loc[spotSamples.index[spotSamples==idPat]],
cell_types=mudata['sc'].obs.cell_subtype2, spot_size=0.015,
legend_fontsize=7)
WARNING: saving figure to file figures/showtest.pdf
[ ]:
3. Co-localization
We computed then co-localization probabilities from the cell type probability matrices. Here we got concatenated co-localization sample matrices of cell type x cell type.
Then we reshaped the co-localization data into a matrix of cell type pairs x samples.
(you can have a closer look at these steps in the preprocessing tutorial)
[11]:
colocPerSample=pd.read_csv('colocPerSample.csv', index_col=0)
colocPerSample.head()
[11]:
| Fib1_SCARA5-Fib1_SCARA5 | Fib1_SCARA5-damaged_CM | Fib1_SCARA5-Capillary_Endo | Fib1_SCARA5-LYVE_FOLR_Macrophages | Fib1_SCARA5-Fib3_C7 | Fib1_SCARA5-healthy_CM | Fib1_SCARA5-Fib2_Myofib | Fib1_SCARA5-Endocardial_Endo | Fib1_SCARA5-Arterial_Endo | Fib1_SCARA5-Neuronal | ... | NK_T-CCL18_Macrophages | NK_T-perivascular_fibroblasts | NK_T-CD_4 | NK_T-vSMCs_2 | NK_T-Lymphatic_Endo | NK_T-NK | NK_T-CD_8 | NK_T-Purkinje_fibers | NK_T-Adipo | NK_T-NK_T | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| control_P17 | 0.017603 | 0.000308 | 0.000992 | 0.000251 | 0.007062 | 0.002586 | 0.004724 | 0.000943 | 0.000412 | 0.000351 | ... | 2.290066e-15 | 0.0 | 3.915381e-05 | 0.0 | 0.0 | 4.538656e-08 | 4.556003e-08 | 0.0 | 0.000000e+00 | 0.000268 |
| RZ_P9 | 0.009307 | 0.000429 | 0.000738 | 0.000003 | 0.005204 | 0.001439 | 0.001625 | 0.000065 | 0.000168 | 0.000046 | ... | 0.000000e+00 | 0.0 | 4.640548e-05 | 0.0 | 0.0 | 9.954633e-05 | 1.643486e-05 | 0.0 | 0.000000e+00 | 0.000784 |
| IZ_P15 | 0.030351 | 0.000000 | 0.000027 | 0.000186 | 0.001200 | 0.000000 | 0.003112 | 0.000072 | 0.000062 | 0.000000 | ... | 0.000000e+00 | 0.0 | 0.000000e+00 | 0.0 | 0.0 | 0.000000e+00 | 0.000000e+00 | 0.0 | 0.000000e+00 | 0.000000 |
| RZ_P6 | 0.040470 | 0.000441 | 0.002752 | 0.000361 | 0.008687 | 0.002928 | 0.007878 | 0.000176 | 0.001022 | 0.001170 | ... | 0.000000e+00 | 0.0 | 7.998369e-25 | 0.0 | 0.0 | 8.593925e-28 | 0.000000e+00 | 0.0 | 0.000000e+00 | 0.000438 |
| RZ_BZ_P3 | 0.021508 | 0.000292 | 0.000567 | 0.000057 | 0.002408 | 0.000483 | 0.006635 | 0.000123 | 0.000052 | 0.000052 | ... | 0.000000e+00 | 0.0 | 0.000000e+00 | 0.0 | 0.0 | 8.585311e-06 | 0.000000e+00 | 0.0 | 7.294563e-35 | 0.000897 |
5 rows × 1089 columns
The sum of the probabilities of every cell type pair in a sample must be = 1
[12]:
colocPerSample.sum(axis=1)
[12]:
control_P17 1.0
RZ_P9 1.0
IZ_P15 1.0
RZ_P6 1.0
RZ_BZ_P3 1.0
FZ_P14 1.0
RZ_BZ_P12 1.0
FZ_GT_P4 1.0
GT_IZ_P13 1.0
GT_IZ_P15 1.0
FZ_P20 1.0
RZ_FZ_P5 1.0
GT_IZ_P9 1.0
RZ_P3 1.0
FZ_GT_P19 1.0
FZ_P18 1.0
IZ_P10 1.0
control_P7 1.0
RZ_P11 1.0
control_P1 1.0
RZ_BZ_P2 1.0
control_P8 1.0
RZ_GT_P2 1.0
dtype: float64
Same cell type interactions will be excluded later on, so we’ll have a list of same cell type interaction pairs in order to subset the co-localization table we’ll generate in the next step.
[13]:
cell_types=CTprops.columns
oneCTints=cell_types+'-'+cell_types
Conditions
We will need the following metadata to subset the samples in control (myogenic) and disease (ischemic):
[14]:
sampleTypesDF=pd.read_csv('MI_sampleTypesDF.csv')
sampleTypesDF.head()
[14]:
| Unnamed: 0 | sample | sampleType | |
|---|---|---|---|
| 0 | 0 | control_P17 | myogenic |
| 1 | 1 | RZ_P9 | remote |
| 2 | 2 | IZ_P15 | ischemic |
| 3 | 3 | RZ_P6 | remote |
| 4 | 4 | RZ_BZ_P3 | border |
4. Differential co-localization analysis
We will test differential co-localization between myogenic and ischemic samples using Wilcoxon rank sums tests:
Null Hypothesis (H0): The median of the population of differences between co-localization probabilities of cell types a and b in myogenic and ischemic samples is zero.
Alternative Hypothesis (H1): The median of the population of differences between co-localization probabilities of cell types a and b in myogenic and ischemic samples is not equal to zero.
[15]:
myo_iscDF=nichesphere.coloc.diffColoc_test(coloc_pair_sample=colocPerSample,
sampleTypes=sampleTypesDF,
exp_condition='ischemic',
ctrl_condition='myogenic')
myo_iscDF.head()
[15]:
| pairs | statistic | p-value | |
|---|---|---|---|
| pairs | |||
| Fib1_SCARA5-Fib1_SCARA5 | Fib1_SCARA5-Fib1_SCARA5 | 0.489898 | 0.624206 |
| Fib1_SCARA5-damaged_CM | Fib1_SCARA5-damaged_CM | -2.44949 | 0.014306 |
| Fib1_SCARA5-Capillary_Endo | Fib1_SCARA5-Capillary_Endo | -2.204541 | 0.027486 |
| Fib1_SCARA5-LYVE_FOLR_Macrophages | Fib1_SCARA5-LYVE_FOLR_Macrophages | -0.489898 | 0.624206 |
| Fib1_SCARA5-Fib3_C7 | Fib1_SCARA5-Fib3_C7 | -2.44949 | 0.014306 |
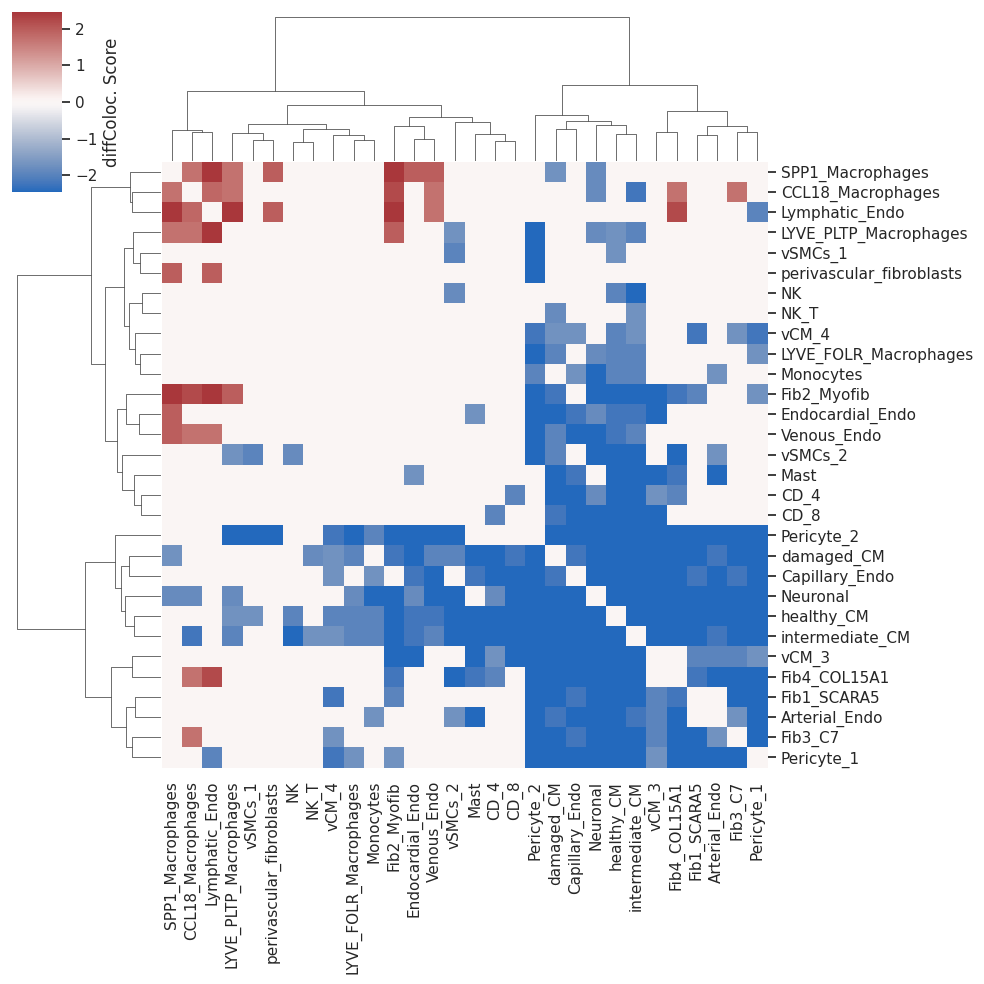
Then we will reshape the data to visualize the Wilcoxon test scores in a heatmap and filter non significant co-localization differences using the parameter p (in this case, scores with p-values > 0.1 are filtered out)
[16]:
myo_isc_HM=nichesphere.tl.pval_filtered_HMdf(testDF=myo_iscDF,
oneCTinteractions=oneCTints,
p=0.1, #threshold p-value to filter
cell_types=cell_types)
myo_isc_HM.head()
[16]:
| Fib1_SCARA5 | damaged_CM | Capillary_Endo | LYVE_FOLR_Macrophages | Fib3_C7 | healthy_CM | Fib2_Myofib | Endocardial_Endo | Arterial_Endo | Neuronal | ... | CCL18_Macrophages | perivascular_fibroblasts | CD_4 | vSMCs_2 | Lymphatic_Endo | NK | CD_8 | Purkinje_fibers | Adipo | NK_T | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fib1_SCARA5 | 0.000000 | -2.449490 | -2.204541 | 0.000000 | -2.449490 | -2.449490 | -1.959592 | 0.000000 | 0.000000 | -2.449490 | ... | 0.000000 | 0.0 | 0.00000 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0 | 0 | 0.000000 |
| damaged_CM | -2.449490 | 0.000000 | -2.204541 | -1.959592 | -2.449490 | -2.449490 | -2.204541 | -2.449490 | -2.204541 | -2.449490 | ... | 0.000000 | 0.0 | -2.44949 | -1.959592 | 0.0 | 0.0 | -2.204541 | 0 | 0 | -1.837117 |
| Capillary_Endo | -2.204541 | -2.204541 | 0.000000 | 0.000000 | -2.204541 | -2.449490 | 0.000000 | -2.204541 | -2.449490 | -2.449490 | ... | 0.000000 | 0.0 | -2.44949 | 0.000000 | 0.0 | 0.0 | -2.449490 | 0 | 0 | 0.000000 |
| LYVE_FOLR_Macrophages | 0.000000 | -1.959592 | 0.000000 | 0.000000 | 0.000000 | -1.959592 | 0.000000 | 0.000000 | 0.000000 | -1.837117 | ... | 0.000000 | 0.0 | 0.00000 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0 | 0 | 0.000000 |
| Fib3_C7 | -2.449490 | -2.449490 | -2.204541 | 0.000000 | 0.000000 | -2.449490 | 0.000000 | 0.000000 | -1.714643 | -2.449490 | ... | 1.714643 | 0.0 | 0.00000 | 0.000000 | 0.0 | 0.0 | 0.000000 | 0 | 0 | 0.000000 |
5 rows × 33 columns
As the cells classified as proliferating cells (prolif) are many different cell types and thus hard to interpret, we’ll remove them for further analysis.
[17]:
myo_isc_HM=myo_isc_HM.loc[myo_isc_HM.columns.str.contains('prolif')==False,myo_isc_HM.index.str.contains('prolif')==False]
We will also remove cells with no significant co-localization differences
[18]:
myo_isc_HM=myo_isc_HM.loc[myo_isc_HM.sum()!=0,myo_isc_HM.sum()!=0]
Now we can plot the differential co-localization scores heatmap
[19]:
sns.set(font_scale=1)
plot=sns.clustermap(myo_isc_HM, cmap='vlag', center=0, method='ward', cbar_kws={'label': 'diffColoc. Score'})
Differential co-localization network
To build the differential co-localization network, we will get an adjacency matrix (adj) based on the euclidean distances among the distributions of significant differential co-localization scores for the different cell types
[20]:
HMdist=pd.DataFrame(scipy.spatial.distance.squareform(scipy.spatial.distance.pdist(myo_isc_HM)),
columns=myo_isc_HM.columns, index=myo_isc_HM.index)
HMsimm=1-HMdist/HMdist.max().max()
##Cell pairs with not significant differential co-localization get 0
HMsimm[myo_isc_HM==0]=0
A cell group dictionary should be used here to visualize different cell groups in different colors. As we don’t have cell groups yet, we’ll have a dictionary of all cells in one group and a list of one color
[21]:
niches_dict={'1_': list(myo_isc_HM.index)}
clist=['#4daf4a']
Now we can plot the differential co-localization network using the colocNW function from NicheSphere. This function has many parameters that can be tuned:
nodeSize for example, defines how the size of the nodes will be calculated. Options are ‘betweeness’, ‘pagerank’ (both network statistics) and None (all nodes have the same size). alpha indicates the transparency of the edges and in goes from 0 (completely transparent) to 1 (opaque) fsize is the size of the figure (x,y)
This function returns the network with the edge weights corresponding to the diff. coloc. scores (positive and negative)
[22]:
plt.rcParams['axes.facecolor'] = "None"
nichesphere.coloc.colocNW(x_diff=myo_isc_HM, #differential co-localization matrix
adj=HMsimm, #adjacency matrix
cell_group=niches_dict,
clist=clist,
nodeSize='betweeness',
lab_spacing=9, #space between node and label
alpha=0.4, #edges transparency
fsize=(12,12)) #figure size
[22]:
<networkx.classes.graph.Graph at 0x15536a1aa190>
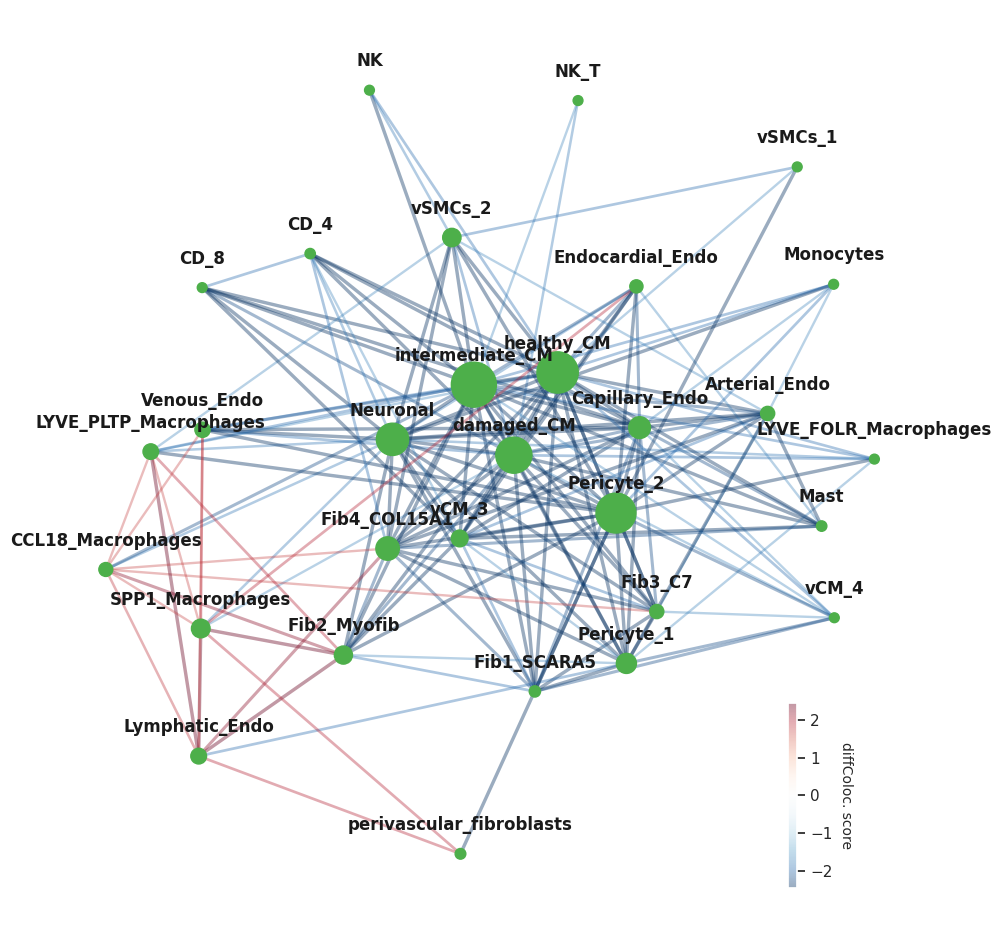
Now we’ll do community detection using Louvain. First we will get the network from the adjacency matrix as we won’t use the signed weights for this
[23]:
gCol_unsigned=nx.from_pandas_adjacency(HMsimm, create_using=nx.Graph)
We will use the community-layout library function CommunityLayout to show the communities in a layout suited for this. This function is compatible with networkx (Hagberg et. al., 2008) community detection functions, which will be used internally as indicated by the parameters community_algorithm and community_kwargs
[24]:
## Calculate community layout
cl=CommunityLayout(gCol_unsigned,
community_compression = 0.4,
layout_algorithm = nx.spring_layout,
layout_kwargs = {"k":75, "iterations":1000},
community_algorithm = nx.algorithms.community.louvain_communities,
community_kwargs = {"resolution":1.1, 'seed':12, 'weight':'weight'})
Building meta-graph
Metagraph is a Graph with 4 nodes and 6 edges
100%|██████████| 4/4 [00:00<00:00, 250.61it/s]
We can extract the communities (niches) as follows:
[25]:
d = {index: list(value) for index, value in enumerate(cl.communities())}
print(pd.DataFrame.from_dict(d, orient='index').T.to_string(index=False))
0 1 2 3
Fib1_SCARA5 Neuronal healthy_CM perivascular_fibroblasts
Pericyte_2 intermediate_CM Arterial_Endo CCL18_Macrophages
vCM_4 CD_8 vSMCs_1 Lymphatic_Endo
Pericyte_1 NK_T NK LYVE_PLTP_Macrophages
Fib4_COL15A1 Capillary_Endo vSMCs_2 Fib2_Myofib
Fib3_C7 Endocardial_Endo Monocytes SPP1_Macrophages
None vCM_3 None Venous_Endo
None LYVE_FOLR_Macrophages None None
None damaged_CM None None
None Mast None None
None CD_4 None None
And then name them
[26]:
niche_names=['1_Stromal', '2_Stressed_CM', '3_Healthy_CM', '4_Fibrotic']
niches_dict=dict(zip(niche_names,list(d.values())))
print(pd.DataFrame.from_dict(niches_dict, orient='index').T.to_string(index=False))
1_Stromal 2_Stressed_CM 3_Healthy_CM 4_Fibrotic
Fib1_SCARA5 Neuronal healthy_CM perivascular_fibroblasts
Pericyte_2 intermediate_CM Arterial_Endo CCL18_Macrophages
vCM_4 CD_8 vSMCs_1 Lymphatic_Endo
Pericyte_1 NK_T NK LYVE_PLTP_Macrophages
Fib4_COL15A1 Capillary_Endo vSMCs_2 Fib2_Myofib
Fib3_C7 Endocardial_Endo Monocytes SPP1_Macrophages
None vCM_3 None Venous_Endo
None LYVE_FOLR_Macrophages None None
None damaged_CM None None
None Mast None None
None CD_4 None None
And assign them colors to color the network nodes according to their niche
[27]:
clist=['#4daf4a', '#0072B5', '#BC3C29', '#ffff33']
niche_cols=pd.Series(clist, index=list(niches_dict.keys()))
niches_df=nichesphere.tl.cells_niche_colors(CTs=CTprops.columns,
niche_colors=niche_cols,
niche_dict=niches_dict)
niches_df.head()
[27]:
| cell | niche | color | |
|---|---|---|---|
| cell | |||
| Fib1_SCARA5 | Fib1_SCARA5 | 1_Stromal | #4daf4a |
| damaged_CM | damaged_CM | 2_Stressed_CM | #0072B5 |
| Capillary_Endo | Capillary_Endo | 2_Stressed_CM | #0072B5 |
| LYVE_FOLR_Macrophages | LYVE_FOLR_Macrophages | 2_Stressed_CM | #0072B5 |
| Fib3_C7 | Fib3_C7 | 1_Stromal | #4daf4a |
Then we can get the node positions to input them to the NicheSphere colocNW function through the pos parameter
[28]:
pos=cl.full_positions
And plot the niches on the community layout
[29]:
plt.rcParams['axes.facecolor'] = "None"
gCol=nichesphere.coloc.colocNW(x_diff=myo_isc_HM,
adj=HMsimm,
cell_group=niches_dict,
clist=clist,
nodeSize='betweeness',
layout=None, #layout needs to be set to None if we provide node positions
lab_spacing=0.05,
thr=1,
alpha=0.4,
fsize=(10,10),
pos=pos, #node positions (from the CommunityLayout function)
edge_scale=1, #edge width
legend_ax=[0.7, 0.05, 0.15, 0.2]) #legend position
#Legend
legend_elements1=[plt.Line2D([0], [0], marker="o" ,color='w', markerfacecolor=clist[i], lw=4,
label=list(niches_dict.keys())[i], ms=10) for i in range(len(list(niches_dict.keys())))]
plt.gca().add_artist(plt.legend(handles=legend_elements1,loc='lower left', fontsize=13, title='Niches',
alignment='left'))
#plt.savefig('diffColocNW_CD.pdf')
[29]:
<matplotlib.legend.Legend at 0x1553693d1e90>
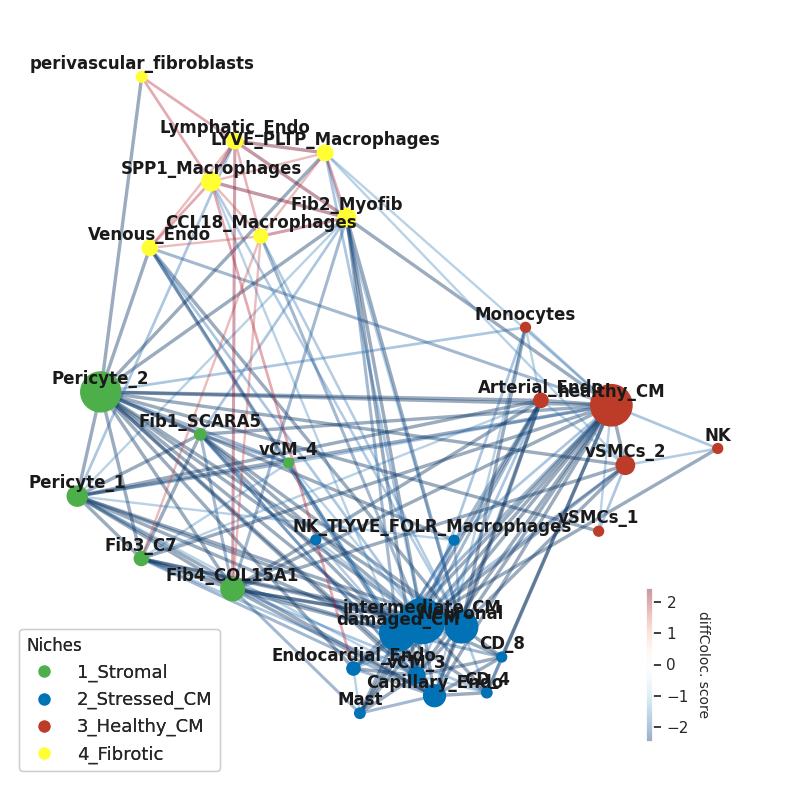
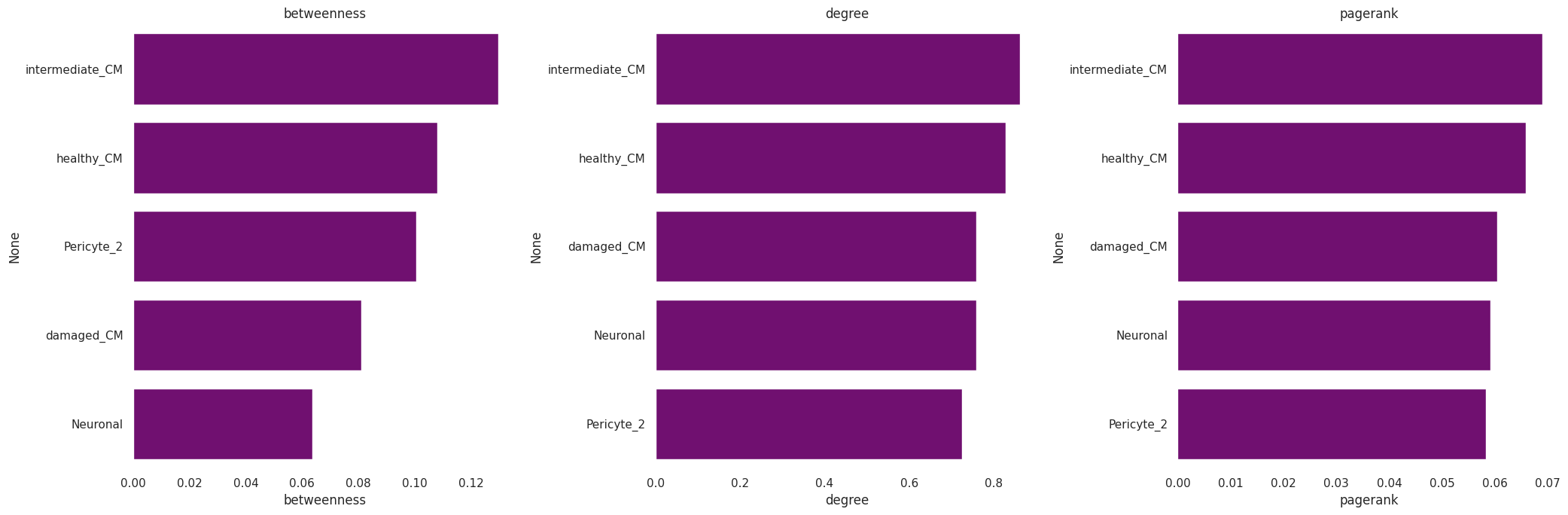
We can also calculate some network statistics with the networkx package functions (this will be done on the signed network):
[30]:
t1=pd.DataFrame({'betweenness':[nx.betweenness_centrality(gCol)[x] for x in list(gCol.nodes)],
'degree':[nx.degree_centrality(gCol)[x] for x in list(gCol.nodes)],
'pagerank':[nx.pagerank(gCol, weight=None)[x] for x in list(gCol.nodes)]})
t1.index=list(gCol.nodes)
[31]:
fig, axes = plt.subplots(1, 3, figsize=(21, 7))
for i in range(len(t1.columns)):
_ = sns.barplot(ax=axes[i], y=t1.sort_values(t1.columns[i], ascending=False).index[0:5], x=t1.columns[i],
data=t1.sort_values(t1.columns[i], ascending=False)[0:5], color='purple')
axes[i].set_title(t1.columns[i])
fig.tight_layout()
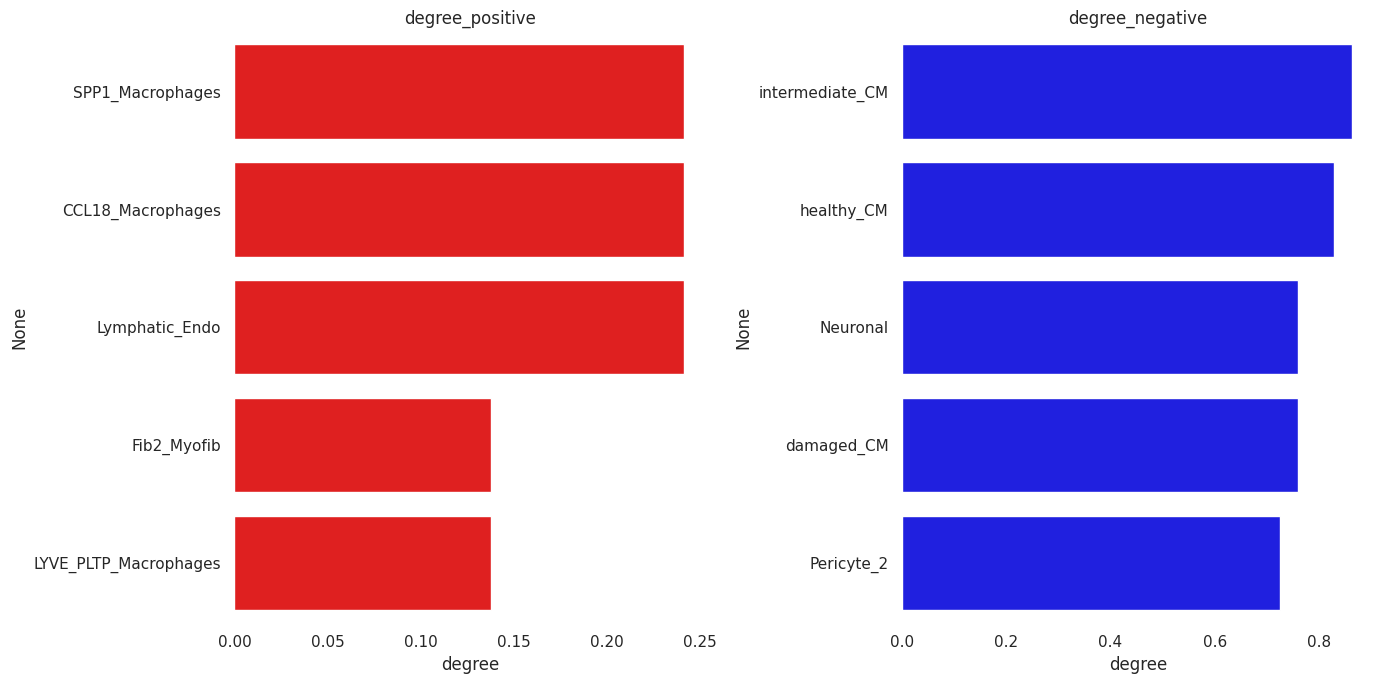
So we can look separately at positive and negative degree:
[32]:
## Positive edges stats
G_pos=gCol.copy()
to_remove=[(a,b) for a, b, attrs in G_pos.edges(data=True) if attrs["weight"] <= 0]
G_pos.remove_edges_from(to_remove)
t1=pd.DataFrame({'degree':[nx.degree_centrality(G_pos)[x] for x in list(G_pos.nodes)]})
t1.index=list(G_pos.nodes)
[33]:
## Negative edges stats
G_neg=gCol.copy()
to_remove=[(a,b) for a, b, attrs in G_neg.edges(data=True) if attrs["weight"] >= 0]
G_neg.remove_edges_from(to_remove)
t2=pd.DataFrame({'degree':[nx.degree_centrality(G_neg)[x] for x in list(G_neg.nodes)]})
t2.index=list(G_neg.nodes)
[34]:
fig, axes = plt.subplots(1, 2, figsize=(14, 7))
_=sns.barplot(ax=axes[0], y=t1.sort_values('degree', ascending=False).index[0:5], x='degree',
data=t1.sort_values('degree', ascending=False)[0:5], color='red')
axes[0].set_title('degree_positive')
_=sns.barplot(ax=axes[1], y=t2.sort_values('degree', ascending=False).index[0:5], x='degree',
data=t2.sort_values('degree', ascending=False)[0:5], color='blue')
axes[1].set_title('degree_negative')
fig.tight_layout()
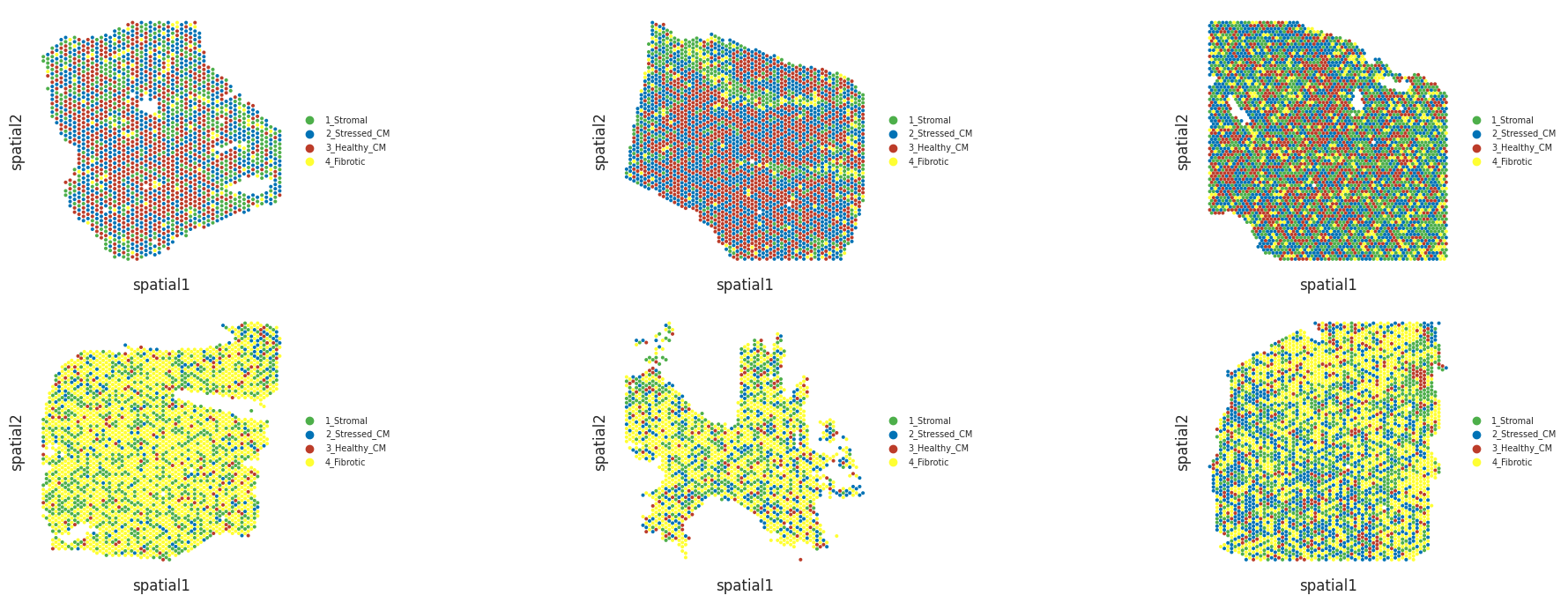
And also visualize niches in slices (spots are colored by the niche to which the cell type with highest proportion belongs)
Niche plots
Now we will visualize the niches in the slices coloring the Visium spots according to the niche of the cell type with the highest proportion.
These are a couple myogenic slices, which will be at the top panels of the next figure:
[35]:
fig, axes = plt.subplots(2, 3, figsize=(21, 7))
plt.close(fig)
for idu,smpl in enumerate(list(sampleTypesDF['sample'][sampleTypesDF['sampleType']=='myogenic'][0:3])):
_ = nichesphere.coloc.spatialNichePlot(adata=mudata['visium'][mudata['visium'].obs.patient_region_id==smpl].copy(),
CTprobs=CTprops.loc[spotSamples.index[spotSamples==smpl]], #dataframe of cell type probabilities per spot
niche_dict=niches_dict,
spot_size=0.015,
niche_colors=niche_cols, #series of colors with niche names as indexes
legend_fontsize=7, save_name='_'+smpl+'.pdf',ax=axes[0][idu])
WARNING: saving figure to file figures/show_control_P17.pdf
<Figure size 640x480 with 0 Axes>
WARNING: saving figure to file figures/show_control_P7.pdf
<Figure size 640x480 with 0 Axes>
WARNING: saving figure to file figures/show_control_P1.pdf
<Figure size 640x480 with 0 Axes>
And a couple ischemic slices, which will be at the bottom panels of the next figure:
[36]:
for idu,smpl in enumerate(list(sampleTypesDF['sample'][sampleTypesDF['sampleType']=='ischemic'][0:3])):
_ = nichesphere.coloc.spatialNichePlot(adata=mudata['visium'][mudata['visium'].obs.patient_region_id==smpl].copy(),
CTprobs=CTprops.loc[spotSamples.index[spotSamples==smpl]],
niche_dict=niches_dict,
spot_size=0.015,
niche_colors=niche_cols,
legend_fontsize=7,
save_name='_'+smpl+'.pdf',ax=axes[1][idu])
WARNING: saving figure to file figures/show_IZ_P15.pdf
<Figure size 640x480 with 0 Axes>
WARNING: saving figure to file figures/show_GT_IZ_P13.pdf
<Figure size 640x480 with 0 Axes>
WARNING: saving figure to file figures/show_GT_IZ_P15.pdf
<Figure size 640x480 with 0 Axes>
[37]:
fig.tight_layout()
fig
[37]:
For further analysis, like differential communication: https://nichesphere.readthedocs.io/en/latest/tutorials.html
, we will need the correspondence data between cell pairs and niche pairs
[38]:
pairCatDFdir=nichesphere.tl.get_pairCatDFdir(niches_df)
pairCatDFdir.to_csv('pairCatDFdir_MIvisium_louvain.csv')
pairCatDFdir.head()
[38]:
| cell_pairs | niche_pairs | |
|---|---|---|
| 0 | Fib1_SCARA5->Fib1_SCARA5 | 1_Stromal->1_Stromal |
| 1 | Fib1_SCARA5->damaged_CM | 1_Stromal->2_Stressed_CM |
| 2 | Fib1_SCARA5->Capillary_Endo | 1_Stromal->2_Stressed_CM |
| 3 | Fib1_SCARA5->LYVE_FOLR_Macrophages | 1_Stromal->2_Stressed_CM |
| 4 | Fib1_SCARA5->Fib3_C7 | 1_Stromal->1_Stromal |
We will also need a filtering object (colocFilt) indicating which cell pairs are differentially co-localized to filter the communication data
[39]:
## Get data of cells present in the adjacency matrix
pairCatDF_filter=[(pairCatDFdir.cell_pairs.str.split('->')[i][0] in HMsimm.index)&
(pairCatDFdir.cell_pairs.str.split('->')[i][1] in HMsimm.index) for i in pairCatDFdir.index]
pairCatDFdir_filt=pairCatDFdir[pairCatDF_filter]
oneCTints_filt=oneCTints[[i.split('-')[0] in HMsimm.index for i in oneCTints]]
[40]:
## Get data to flag differentially co-localized cell pairs in the adjacency matrix
colocFilt=nichesphere.tl.getColocFilter(pairCatDF=pairCatDFdir_filt,
adj=HMsimm,
oneCTints=oneCTints_filt.str.replace('-', '->'))
colocFilt.to_csv('colocFilt_MIvisium_louvain.csv')
colocFilt.head()
[40]:
| filter | |
|---|---|
| cell_pairs | |
| Fib1_SCARA5->Fib1_SCARA5 | 1.0 |
| Fib1_SCARA5->damaged_CM | 1.0 |
| Fib1_SCARA5->Capillary_Endo | 1.0 |
| Fib1_SCARA5->LYVE_FOLR_Macrophages | 0.0 |
| Fib1_SCARA5->Fib3_C7 | 1.0 |
We will need the niche - cell type - color correspondence data, the co-localization network and nodes positions for further analysis as well
[41]:
niches_df.to_csv('niches_df_MIvisium_louvain.csv')
nx.write_graphml_lxml(gCol, "colocNW_MIvisium_louvain.graphml")
np.save('colocNW_pos.npy', pos)
[ ]: